
Болезнь Фабри (БФ) – это редкое наследственное заболевание, которое относится к лизосомным болезням накопления и обусловлено мутациями гена GLA, расположенного на Х-хромосоме и кодирующего синтез лизосомного фермента α-галактозидазы А [1]. Снижение или полное отсутствие активности этого фермента приводит к накоплению гликосфинголипидов в клетках различных органов и тканей, в т.ч. почек, сердца, центральной и периферической нервных систем, что приводит к нарушению их функции и развитию жизнеугрожающих осложнений, таких как диализ-зависимая хроническая почечная недостаточность, нарушения ритма сердца, сердечная недостаточность и инсульт [2]. При классическом варианте БФ первые симптомы (невропатическая болезнь, ангиокератомы, сниженное потоотделение) появляются в детском возрасте, а в возрасте 20–40 лет развивается поражение внутренних органов, которое постепенно прогрессирует и может приводить к инвалидизации или смерти в трудоспособном возрасте [3]. Выделяют также поздний, или атипичный, вариант заболевания, который характеризуется отсутствием ранних симптомов, поздним началом, обычно в возрасте 40–50 лет, и изолированным или преимущественным поражением одного органа, например сердца или почек [4].
Основа лечения БФ – применение рекомбинантных препаратов a-галактозидазы А (агалсидаза альфа и агалсидаза бета), которые вводят внутривенно каждые 2 недели. В соответствии с клиническими рекомендациями, одобренными Минздравом Российской Федерации (РФ) в 2019 г., лицам мужского пола с классическим вариантом БФ ферментозаместительную терапию (ФЗТ) целесообразно начинать сразу после установления диагноза БФ, в то время как женщинам ее назначают только при наличии невропатической боли или признаков поражения почек, сердца или головного мозга, т.е., как правило, в старшем возрасте [5]. Это связано с особенностями течения Х-сцепленного наследственного заболевания у женщин, у которых мутантный ген находится в гетерозиготном положении (а не в гемизиготном как у мужчин), а поражение внутренних органов развивается позднее и в целом прогрессирует медленнее, чем у мужчин [3]. Тем не менее женский пол не исключает возможности тяжелого течения БФ. В российской популяции поражение почек, сердца и/или головного мозга было выявлено у 83% из 72 взрослых пациенток с БФ, а по крайней мере один неблагоприятный исход заболевания в возрасте от 19 до 70 лет (медиана – 44 года) зарегистрировали в 25% случаев [6].
В РФ зарегистрированы оригинальные рекомбинантные препараты a-галактозидазы А – агалсидаза альфа (Реплагал®) и агалсидаза бета (Фабразим®), которые назначают в дозах 0,2 и 1,0 мг/кг соответственно. В 2014 г. в Республике Корея на основании серии доклинических и клинических исследований был одобрен для клинического применения биоаналог агалсидазы бета – ISU303, разработанный корейской компанией ISU ABXIS Co., Ltd. (торговое наименование – Фабагал®). Биоэквивалентность препарата ISU303 и оригинальной агалсидазы бета была установлена в клиническом исследовании I фазы у 18 здоровых добровольцев [7]. Компания ООО «НПО Петровакс Фарм» получила права на проведение клинических исследований препарата ISU303, его регистрацию и вывод на рынок на территории Евразийского Экономического Союза (ЕАЭС).
Целью двойного слепого рандомизированного исследования I фазы, проведенного в рамках регистрации препарата ISU303 на территории ЕАЭС, было сравнение фармакокинетики, переносимости и безопасности биоаналога агалсидазы бета (Фабагал®) и оригинального препарата (Фабразим®) после однократного введения здоровым добровольцам.
Материал и методы
Двойное слепое рандомизированное исследование I фазы с активным контролем в параллельных группах было проведено в одном исследовательском центре в соответствии с принципами Хельсинкской декларации Всемирной медицинской ассоциации, правилами надлежащей клинической практики Международного совета по гармонизации (ICH GCP) и Евразийской экономической комиссии и национальными регуляторными требованиями. Протокол исследования был одобрен локальным этическим комитетом Исследовательского центра и Советом по этике при Минздраве РФ. Все добровольцы подписали информированное согласие на участие в исследовании. Спонсор исследования (ООО «НПО Петровакс Фарм», Россия) предоставил исследуемый препарат и препарат сравнения, но не принимал непосредственного участия в проведении исследования и статистическом анализе полученных данных.
Исследуемый препарат – Фабагал® (ISU303, агалсидаза бета, лиофилизат для приготовления концентрата для приготовления раствора для инфузий, 35 мг, ООО «НПО Петровакс Фарм», Россия/ISU ABXIS Co., Ltd., Республика Корея); препарат сравнения – Фабразим® (агалсидаза бета, лиофилизат для приготовления концентрата для приготовления раствора для инфузий, 35 мг, Джензайм Европа Б.В., Нидерланды).
В исследование включили здоровых добровольцев мужского пола в возрасте 18–45 лет, соответствовавших критериям отбора. Критерии включения: подписанное информированное согласие, индекс массы тела (ИМТ) – 18,5–25,0 кг/м2, масса тела – 50–75 кг, отсутствие признаков заболевания по данным стандартных клинических, лабораторных и инструментальных методов обследования, личного и семейного анамнеза, согласие пользоваться надежными методами контрацепции на протяжении всего исследования.
Критерии невключения: непереносимость любого компонента исследуемых препаратов или препаратов для премедикации (ибупрофен, дифенгидрамин), тяжелые инфузионные реакции или клинически значимая гиперчувствительность к любым лекарственным препаратам или продуктам питания в анамнезе, систолическое артериальное давление (АД) ниже 100 или выше 130 мм рт.ст., диастолическое АД ниже 70 или выше 85 мм рт.ст., частота сердечных сокращений менее 60 или более 80 в минуту, острые инфекционные заболевания в течение 4 недель до скрининга, курение, употребление по крайней мере 10 единиц алкоголя в неделю, злоупотребление алкоголем, наркотическими веществами или лекарственными препаратами в анамнезе, употребление стимулирующих напитков в больших объемах, положительный результат анализа мочи на наличие наркотических и сильнодействующих веществ, положительный тест на содержание паров алкоголя в выдыхаемом воздухе, регулярный прием любых рецептурных и безрецептурных лекарственных препаратов, биоактивных добавок, витаминов менее чем за 2 недели до начала исследования, прием препаратов зверобоя продырявленного (Hypericum perforatum) менее чем за 30 дней до включения в исследование, прием ингибиторов или индукторов микросомальных ферментов печени (барбитураты, омепразол, циметидин и т.д.) или противовирусных препаратов менее чем за 2 месяца до включения в исследование, донорство цельной крови в течение предыдущих 8 недель или компонентов крови в течение предыдущих 4 недель, участие в других клинических исследованиях лекарственных средств в течение предыдущих 3 месяцев, психические, физические и прочие причины, не позволяющие добровольцу выполнять условия протокола исследования.
Исследование состояло из 3 этапов (продолжительность участия одного добровольца – до 24 дней):
- Скрининг (день -14... день 1).
- Визит 1 – введение исследуемого препарата в центре (день 1-й… день 2-й).
- Визит 2 – наблюдение (день 7±1).
Добровольцев, прошедших скрининг, во время визита 1 распределяли в 2 группы в соотношении 1:1 с помощью рандомизационной веб-системы IWRS (Interactive Web Response System). Исследуемые препараты вводили в день 1 утром (~7:00–12:00) натощак, однократно, внутривенно (5-часовая инфузия со скоростью 100 мл/ч в течение 5 часов с помощью инфузомата) в дозе 1 мг/кг. Раствор для введения (500 мл) готовила разослепленная медицинская сестра, не вовлеченная в другие процедуры клинического исследования. Для профилактики возможных инфузионных реакций проводили премедикацию ибупрофеном 200 мг внутрь и дифенгидрамином 50 мг внутрь за 40 минут до инфузии исследуемого препарата. Питание добровольцев на протяжении 3-дневной госпитализации было стандартизированным (день -1: ужин как минимум за 10 часов до введения исследуемого препарата, день 1-й: обед и ужин соответственно через 6 и 12 часов после его введения, день 2-й: завтрак после взятия последнего образца крови). Во время инфузии доброволец мог выпить не более 400 мл воды. За 72 часа до введения препаратов не разрешалось употребление пищевых продуктов или напитков, содержащих кофеин и ксантин (например, кофе, чай, кола и шоколад), и алкогольных напитков.
Кровь для анализа лабораторных показателей брали в 16 точках: перед введением, во время введения (1-й, 3, 5-й час) и после введения (через 5, 10, 20, 30, 45 минут и 1, 1,5, 2, 3, 4, 7, 19 часов). Образцы крови объемом до 4 мл замораживали и направляли в центральную лабораторию.
Количественное определение агалсидазы бета проводилось косвенно по оценке активности α-галактозидазы А спектрофлуориметрическим методом с помощью коммерческого набора Alpha Galactosidase Activity Assay Kit производства Abcam (США). Количественный анализ активности проводили методом флуориметрии (Ex/Em 360/445 нм). Методика анализа была валидирована по следующим параметрам: специфичность, селективность, градуировочная кривая (линейность), точность, прецизионность, нижний предел количественного определения, перенос пробы, стабильность, эффект разбавления, минимально необходимое разведение. Подтвержденный аналитический диапазон методики составил 50–1000,0 нг/мл для агалсидазы бета в плазме крови.
Первичным критерием оценки биоэквивалентности была площадь под кривой «плазменная концентрация–время» от нулевой точки до бесконечности (AUC0–∞), вторичными – площадь под кривой «плазменная концентрация–время» от нулевой точки до времени t (AUC0–t) и максимальная плазменная концентрация (Cmax). Измеряли также tmax – время достижения Cmax, t1/2 – период полувыведения из плазмы, Kel – константу элиминации, относящуюся к терминальной фазе, CL – клиренс, Vd – объем распределения.
Безопасность изучаемых препаратов оценивали на основании частоты и характера побочных эффектов (до дня 7±1) и серьезных побочных эффектов (до дня 30), жизненно важных показателей, результатов физического обследования, электрокардиограммы (ЭКГ), биохимического (глюкоза, общий белок, альбумины, печеночные ферменты, общий билирубин, общий холестерин, креатинин, мочевина, С-реактивный белок, калий, натрий, кальций, магний, хлориды и фосфаты), клинического анализов крови и общего анализа мочи.
Для статистического анализа использовали программное обеспечение IBM® SPSS® Statistics Version 26.0. Сравнительный анализ фармакокинетики проводили всем участникам, у которых не было зарегистрировано существенных нарушений протокола и был оценен хотя бы один фармакокинетический параметр. Безопасность оценивали у всех рандомизированных участников, получавших инфузию препарата. Для количественных переменных рассчитывали среднее значение, стандартное отклонение (SD), медиану, первый и третий квартили, минимальное и максимальное значения и число валидных наблюдений (N). Качественные показатели представлены в виде частот и долей в процентах. Для значений концентрации и фармакокинетических параметров рассчитывали также коэффициент вариабельности (%).
Основной фармакокинетический параметр AUC0–∞ сравнивали после логарифмического преобразования с помощью модели дисперсионного анализа ANOVA для параллельного дизайна. Препараты считают эквивалентными по фармакокинетике, если границы двустороннего ДИ для отношения геометрических средних значений AUC0–∞ исследуемого препарата и препарата сравнения (T/R) находятся в пределах 80,00–125,00. Если расчетная мощность теста (с учетом коэффициента вариации) составляет ≥90%, то тестирование гипотезы о фармакокинетической эквивалентности проводится на уровне α=5% с расчетом 90% ДИ. Если мощность оказывается <90%, то тестирование первичной гипотезы проводится на уровне α=3,04% (0,0304) с расчетом двустороннего 93,92% ДИ.
Расчетный размер выборки исследования составил 52 человека. В связи с ограниченным объемом данных о вариабельности основного фармакокинетического параметра в исследовании был запланирован разослепленный промежуточный анализ для определения достаточности запланированного размера выборки для подтверждения биоэквивалентности препаратов сравнения. По результатам промежуточного анализа могло быть принято решение об остановке исследования или дополнительном наборе участников (максимум до 104).
Результаты
В отборочном этапе приняли участие 63 добровольца. Одиннадцать из них выбыли до рандомизации, а оставшиеся 52 добровольца были рандомизированы в 2 группы по 26 человек. Все они получали исследуемый препарат или препарат сравнения и завершили исследование в соответствии с протоколом. Отклонения от протокола, которые могли бы повлиять на результаты фармакокинетического анализа, зарегистрированы не были.
Все добровольцы были мужчинами европеоидной расы в возрасте от 20 до 37 лет с ИМТ от 18,5 до 24,9 кг/м2. Две группы были сопоставимыми по демографическим и другим исходным показателям, в частности, средний возраст участников в основной группе составил 27,8±4,3 года, в группе сравнения – 26,5±5,4 года, а средний ИМТ – 21,9±1,7 и 21,1±1,6 кг/м2 соответственно. Тесты на алкоголь в выдыхаемом воздухе и наркотические вещества и котинин в моче у всех добровольцев были отрицательными.
В выборку для сравнительного анализа фармакокинетики были включены все 52 добровольца. Усредненные фармакокинетические профили (концентрация–время) α-галактозидазы А в плазме крови при применении исследуемого препарата (Фабагал®) и препарата сравнения (Фабразим®) представлены на рисунке, а показатели фармакокинетики – в таблице.
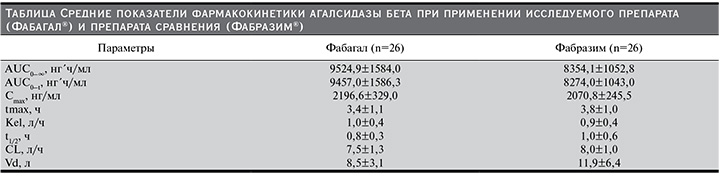
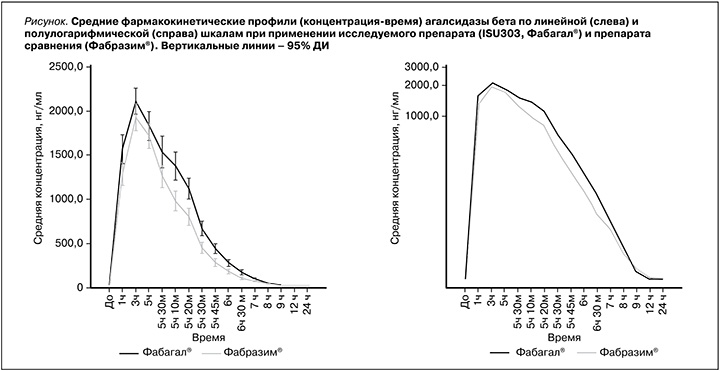
Средняя AUC0–∞, служившая первичным показателем фармакокинетики при сравнительном анализе, при введении исследуемого препарата была незначительно выше, чем при введении Фабразима®: 9524,9±1584,0 и 8354,1±1052,8 нг´ч/мл соответственно (см. таблицу). Межиндивидуальная вариабельность AUC0-∞ α-галактозидазы А, которую оценивали с помощью геометрического коэффициента вариации (gCV, %),
при применении двух препаратов находилась в пределах 12,7–16,7%. Расчетная мощность (83,9%) оказалась ниже 90%, поэтому первичную гипотезу тестировали на скорректированном уровне α=3,04%. Двусторонний 93,92% ДИ для отношения средних геометрических значений AUC0–∞ агалсидазы бета при введении исследуемого препарата и препарата сравнения составил 104,99–122,45% и находился в допустимых пределах 80,00–125,00%, что подтверждало эквивалентность препаратов Фабагал® и Фабразим®.
Средние значения AUC0–t и Cmax при введении исследуемого препарата также были несколько выше, чем при введении препарата сравнения (см. таблицу). По данным дисперсионного анализа (ANOVA), 90% ДИ для отношения средних геометрических значений AUC0–t и Cmax, рассчитанных на основании показателей концентрации агалсидазы бета в плазме, составили 106,25–121,54 и 99,54–112,16% соответственно и находились в пределах границ биоэквивалентности.
Другие показатели фармакокинетики агалсидазы бета, в т.ч. tmax, Kel, t1/2, CL и Vd, также были сходными при введении двух препаратов. Среди показателей, отражавших элиминацию, средние значения t1/2 и CL были незначительно меньше, а значение Kel незначительно больше в основной группе, что может указывать на более быстрое выведение исследуемого препарата. Средние значения Vd при введении исследуемого препарата и препарата сравнения составили 8,5 и 11,9 л соответственно. Безопасность анализировали у всех рандомизированных участников исследования. Во время исследования не было зарегистрировано побочных эффектов и серьезных побочных эффектов, а также отклонений жизненно важных показателей, результатов физического исследования и ЭКГ и клинически значимых отклонений лабораторных показателей от нормы. Клинически незначимые отклонения лабораторных показателей от нормы встречались в единичных случаях: снижение числа тромбоцитов в крови у 1 (3,8%) участника в группе сравнения и у 2 (7,7%) участников в основной группе, снижение числа ретикулоцитов в крови у 1 (3,8%) участника в группе сравнения и снижение содержания общего белка в крови у 1 (3,8%) участника в группе сравнения.
Обсуждение
Результаты двойного слепого рандомизированного исследования I фазы подтвердили биоэквивалентность биоаналога агалсидазы бета (Фабагал®) и оригинального препарата (Фабразим®) при однократном применении в рекомендуемой дозе 1 мг/кг у здоровых добровольцев. Биоэквивалентность была установлена на основании анализа первичного показателя фармакокинетики агалсидазы бета (AUC0–∞) и подтверждена результатами оценки вторичных показателей (AUC0–t и Cmax). ДИ отношений средних геометрических значений указанных показателей находились в допустимых пределах 80,00–125,00%, установленных для исследований биоэквивалентности. Среди показателей, отражающих элиминацию, средние значения t1/2 и CL были незначительно меньше, а значение Kel незначительно больше при введении исследуемого препарата, что может указывать на незначительно более быстрое его выведение. Выявленные различия средних значений Vd при введении двух препаратов согласуются с имеющимися данными о высокой вариабельности объема распределения агалсидазы бета [8]. Вывод о биоэквивалентности сравниваемых препаратов был сделан на основании результатов предусмотренного протокола промежуточного анализа данных 52 добровольцев, поэтому исследование было завершено на этом этапе, а дополнительный необязательный набор участников, допускавшийся в соответствии с адаптивным дизайном исследования, не проводился. Кроме того, исследование продемонстрировало безопасность однократной инфузии биоаналога агалсидазы бета, которая не сопровождалась какими-либо побочными эффектами или отклонениями результатов физического и лабораторных исследований от нормы.
В рамках нашего исследования не оценивалась иммуногенность, т.к. в плацебо-контролируемом клиническом исследовании фазы I, проводившемся в Республике Корея, у 6 здоровых добровольцев через 30 дней после однократной инъекции препарата ISU303 в дозе 1 мг/кг антитела к α-галактозидазе A обнаружены не были [7]. В японском исследовании через 28 дней после однократного введения здоровым добровольцам другого биоаналога агалсидазы бета (JR-051) или оригинального препарата антитела к α-галактозидазе A также не определялись [9]. Тем не менее на фоне длительной ферментозаместительной терапии агалсидазой бета у пациентов с БФ возможно образование антител к α-галактозидазе A при применении как оригинального препарата [10], так и его биоаналогов [11]. Клиническое значение возможной иммуногенности рекомбинантных препаратов α-галактозидазы A остается неопределенным. B. Benichou et al. в ретроспективном исследовании у 134 пациентов с БФ, получавших агалсидазу бета в дозе 1 мг/кг каждые 2 недели в течение до 5 лет, не выявили корреляции между титрами IgG антител к α-галактозидазе A и развитием клинических исходов, изменением расчетной скорости клубочковой фильтрации (СКФ) или содержания глоботриазоилцерамида (GL3) в плазме, хотя высокие титры антител ассоциировались с отложением GL3 в эндотелиальных клетках капилляров кожи [10]. В то же время в других исследованиях наличие нейтрализующих антител сопровождалось менее выраженным снижением содержания Lyso-GL3 на фоне длительной терапии агалсидазой бета [12]. Необходимо подчеркнуть, что в обычной клинической практике определение антител к α-галактозидазе A во время ФЗТ не проводится с учетом отсутствия стандартизированных и общепринятых методов оценки иммуногенности агалсидазы альфа или агалсидазы бета, что затрудняет и интерпретацию результатов опубликованных исследований [13].
Эффективность и безопасность агалсидазы бета в лечении БФ были установлены еще более двух десятилетий назад в многоцентровом рандомизированном двойном слепом плацебо-контролируемом исследовании, в котором ФЗТ этим препаратом в дозе 1 мг/кг вызывала исчезновение отложений GL3 из эндотелия микрососудов почек, сердца и кожи у 58 пациентов с БФ [14]. D. Germain et al. проанализировали отдаленные результаты ФЗТ (медиана длительности около 10 лет) у 52 пациентов, принимавших участие в этом исследовании [15]. В течение указанного срока у 81% пациентов не было зарегистрировано каких-либо тяжелых клинических исходов, а выживаемость к концу наблюдения составила 94%. ФЗТ была более эффективной в тех случаях, когда ее начинали в более молодом возрасте в отсутствие выраженных признаков поражения почек. Эффективность и безопасность агалсидазы бета подтверждены в многочисленных контролируемых и наблюдательных клинических исследованиях, в т.ч. проводившихся на основании данных международного регистра (Fabry Registry), в который были включены более 8000 пациентов с БФ [16]. По данным систематизированного обзора опубликованных клинических исследований, длительная ФЗТ вызывала снижение содержания GL3 в плазме, моче и клетках различных органов, уменьшение невропатической боли в конечностях и боли в животе, замедляла снижение расчетной СКФ, стабилизировала массу миокарда левого желудочка, улучшала исходы заболевания и показатели качества жизни [17, 18]. Благоприятное влияние ФЗТ рекомбинантными препаратами α-галактозидазы А на невропатическую боль, массу миокарда левого желудочка и функцию почек было продемонстрировано и в российской популяции у 35 взрослых мужчин и женщин с БФ, продолжавших лечение в течение около 5 лет [19].
Эффективность и безопасность биоаналога агалсидазы бета были продемонстрированы в многоцентровом открытом 6-месячном исследовании II фазы, проведенном в Республике Корея [11]. В него были включены 10 взрослых пациентов с БФ, 8 из которых ранее получали лечение оригинальной агалсидазой бета. Содержание GL3 в плазме, которое было первичным критерием эффективности, достоверно снизилось (p<0,01) у всех пациентов, в т.ч. получавших и не получавших ФЗТ ранее, и оставалось стабильным на протяжении всего исследования. Содержание других лабораторных маркеров, в т.ч. GL3 в моче и Lyso-GL3 в плазме, имело тенденцию к снижению, однако эти изменения не достигли статистической значимости. Толщина стенки левого желудочка и межжелудочковой перегородки, СКФ, интенсивность невропатической боли и показатели качества жизни после замены оригинального препарата на биоаналог не изменились. Серьезных побочных эффектов зарегистрировано не было.
Таким образом, результаты этого исследования показали, что Фабагал® по эффективности и безопасности не уступает оригинальному препарату агалсидазы бета. В настоящее время планируются дополнительные программы клинического наблюдения пациентов, получающих Фабагал®, в т.ч. в РФ.
Заключение
Результаты двойного слепого рандомизированного исследования I фазы у здоровых добровольцев подтвердили биоэквивалентность биоаналога агалсидазы бета (Фабагал®) и оригинального препарата (Фабразим®) при однократном внутривенном введении в рекомендуемой дозе 1 мг/кг. Переносимость исследуемого препарата была хорошей. Каких-либо побочных эффектов или клинически значимых отклонений результатов физического обследования, ЭКГ и лабораторных показателей от нормы зарегистрировано не было.


